Вдохновение: Высокопроизводительное секвенирование сделало исследование новых моделей органических объектов более разумным. Несмотря на то, что сбор другого генома в любом случае может быть непомерным и хлопотным, вполне возможно использовать RNA-seq для группировки мРНК. Не имея известного генома, важно собрать эти группы заново, принимая во внимание возможные варианты изоформ и уникальный диапазон значений артикуляции.
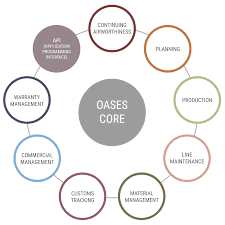
Мы представляем пакет продуктов под названием «Программное обеспечение Oases», предназначенный для эвристического сбора результатов секвенирования РНК без эталонного генома, в широком диапазоне значений артикуляции и при наличии дополнительных изоформ. Это достигается за счет использования различных длин хэшей, динамического отсеивания шума, четкого выбора событий присоединения и умелого объединения разных групп. Он был опробован на данных секвенирования РНК человека и мыши и, по всей видимости, работает, по существу, с агентами, создающими новые транскрипты Trans Abyss и Trinity.
ВВЕДЕНИЕ
Передовое секвенирование сообщающихся мРНК (RNA-seq) постепенно меняет область применения). Первичные попытки найти изоформы сообщаемого качества зависели от планирования секвенирования РНК экзонов и пересечений экзонов с экзонами из известного комментария (Цзянг и Вонг, 2009; и др., 2008; Ричард и др., 2010; Султан и др.). ., 2008; Ван и др., 2008). Впоследствии были созданы основанные на ссылках стратегии инициализации мышц желудка для сбора информации о секвенировании РНК с использованием только механизмов считывания, что вызвало базовый комментарий.
К сожалению, использование эталонного генома вообще невозможно себе представить. Несмотря на снижение стоимости реагентов для секвенирования, полное исследование генома, от изучения до завершения встречи, по-прежнему непомерно и хлопотно. Время от времени рассматриваемая модель адекватно не совпадает с эталонной, поскольку она происходит от альтернативной линии или линии до такой степени, что отображения не являются полностью надежными. В этих случаях использовались новые агенты для конструирования генома для создания групп записей или переносов из секвенирования РНК, просмотра без какого-либо эталонного генома.
Тем не менее, эти кратко читаемые геномные конструирующие агенты, представленные на диаграмме геномных конструирующих агентов (и Birney, 2008; Simpson et al., 2009), вызывают понятные подозрения относительно справедливости включения и расположения. Конечно, глубина включения существенно меняется между записями, изоформами и областями записи, поэтому ее нельзя использовать для определения уникальности областей или для выделения неправильного расположения. Кроме того, эти аппараты приспособлены для доставки длинных прямых содержаний из данной схемы, а не для выделения покрывающих последовательностей, вносимых изоформами одиночного качества. Это влияет на различные усовершенствования, включая исправление ошибок, перефразирование местоположения и использование пар чтения. Соответственно, эти стратегии не подходят для обработки расшифрованной информации, которая не соответствует обоим этим подозрениям.
Тем более, что в последнее время были созданы конвейеры сбора транскриптов для постобработки результатов повторного конструирования генома агентами: Velvet и (Martin et al., 2010; Robertson et al., 2010; и Montoya-Burgos, 2010). ). Обычная идея, разделяемая этими конвейерами, состоит в том, чтобы запустить строительный агент на различных длинах километров и объединить эти собрания в одно. Основанием для этой методологии является объединение более деликатных (меньшие положительные значения k) и более явных собраний (более высокие положительные значения k).
Конвейер, представленный Робертсоном и др. (2010), дополнительно рассматриваются варианты избирательной прививки. Он различает их посредством поиска связанных собраний до такой степени, что они связаны в воздушном кармане товарного знака, и один из них имеет длину ровно (2k−2). Эти воздушные карманы сначала устраняются, а затем добавляются к последним сборкам, чтобы воссоздать заменяющие варианты.
Ряд алгоритмических аналитиков использовали диаграммы прививки для решения выборных объединений, которые имеют непосредственное отношение к диаграммам, на что обратили внимание Хебер и др. (2002). Эта гомология между информационными структурами открывает шанс на появление нового краткого конструирующего агента, как это показано в расчете Тринити. Тринити начинает с жадного расширения, связывания их в диаграмму, а затем, в этой точке, и высвобождения адекватно пройденных путей через эту диаграмму. . Trinity предназначен для воспроизведения глубоко переданных записей на полную длину, используя только одну длину, и вы можете проконсультироваться для получения более подробной информации в лаборатории Baytek.